表面分析装置によるLiイオン電池材料の解析事例
日本電子 News Vol.46 No.6
堤 建一、島 政英、田中 章泰
日本電子(株)SA事業ユニット
1.緒言
オージェ電子分光法(AES: Auger Electron Spectroscopy)とX線光電子分光法(XPS: X-ray Photoelectron Spectroscopy)は、表面から深さ数nmの領域の元素分析を行う手法として広く用いられている。特に、これら2つの分析法は、水素、ヘリウムに次ぐ軽元素であるLiも直接検出することができ、さらに定性的な評価だけでなく定量的な評価および化学結合状態の評価も行うことができるため、古くからLiに関する研究・開発に用いられてきた。しかし現状のLiイオン電池に関する書籍や発表論文等をインターネットの文献検索(http://scholar.google.com/)等で調べてみると、XPSを使った研究成果を含む文献は2000件も報告されているが、同じ検索条件でAESを使った文献を調べてみると500件程度しか該当しなかった。これはXPSとAESのユーザー数の違いによるものもあろうが、AESではLi分析が困難であり、研究に用いることが難しいと誤解されていることも一因であると考えられる。事実、Li KVVのオージェ電子の運動エネルギーは50eVという低エネルギー帯にあり、二次電子の大きなバックグラウンド上に位置し、他の元素のオージェのピークとも重なることもあって、試料によっては、スペクトルを測定しただけではLiのピークの同定すら難しい場合もある。もう一つの理由は、Liオージェ電子の脱出深さが浅いということである。50eVという低い運動エネルギーの電子はわずか1~2nmの厚みのコンタミネーションが付着しても、そのピーク強度は著しく低下し、検出することすら困難となる。一方XPSの場合には、Liから放出される光電子の運動エネルギーは約1200~1400 eVとオージェ電子と比較し非常に高い。このことによってXPSでは試料表面が若干汚染されていても十分にLi由来のスペクトルを観察できることが多い。また他の元素スペクトルとの重なりが小さいこともLiの検出を容易にしている。Liのエネルギー領域は特に遷移金属とスペクトルが重なることが多い。Liイオン電池によく用いられる遷移金属元素にはMn,Fe,Co,Niなどがある。しかしながらXPSの場合にはスペクトルが完全に重なるのはFeだけでありそれ以外では十分に観察できるほどその位置は離れている。つまり、AESでは、試料前処理の段階でコンタミネーションの付着をいかに防ぐかも重要であり、前処理方法を誤ると、同じ試料でもXPSではLiが検出できるがAESでは検出できないということもあり、このような事例もよく見受けられる。こうしてみると、LiはAESによる観察には適さない元素のようであるが、測定者が正しい前処理方法や分析時の注意点を理解すれば、Liそのものの感度はXPSよりも高く、倍率が数万~数10万倍の像に対応したLiマッピングにも他の元素に比べ短時間で測定することが可能である。つまりAES、XPSそれぞれにLiの分析に対して有利・不利な点がある。これらの分析法の特長を理解したうえで使い分けることにより、Liの分析が有用であることを、Liイオン電池材料に注目し分析・解析を行った結果を用いて報告する。
2.表面分析におけるLi検出のための前処理方法
2.1 Liを含む材料の取り扱いの注意点
Liイオン電池材料中のLi元素は、その特性からLiイオンとして隣接する物質(固体・液体にかかわらず)に容易に移動するため、試料前処理を行う場合にはその取り扱いに注意が必要である。
その必要性を示す例として、Liイオン電池材料の一つであるLiCoO2粒子を例に挙げる。清浄な粒子表面でAES分析を行うと、LiピークはFig.1のように微分スペクトルに明確に観察される。しかし、この粒子に対して、表面のコンタミネーションを除去することを意図して、エチルアルコール等の溶媒で超音波洗浄を行うと、洗浄後のLiCoO2粒子表面からはLiピークがほとんど検出されなくなる。これは、粒子表面の領域のLi元素が洗浄時の溶媒であるエチルアルコール中に溶出し、Liの表面濃度が低下したためだと考えられる。Fig.1の場合にはLiピークは消失し、ほぼコバルト酸化物になっていることがわかる。この現象は、水はもちろんのこと、その他の有機溶媒でも生じる可能性は高いので、最表面を分析対象とするAESやXPSでは、特に注意が必要である。AESやXPSで表面分析を行う場合には、特に必要がなければ、無機・有機溶媒による粒子表面の洗浄は行わない方がよい。
もし、表面にコンタミネーションが付着したLi含有粒子を表面分析する場合には、溶媒洗浄せずに、乳鉢と乳棒で粉末同士を擦り合わせて新たな清浄表面を露出させてから、分析することを推奨したい。
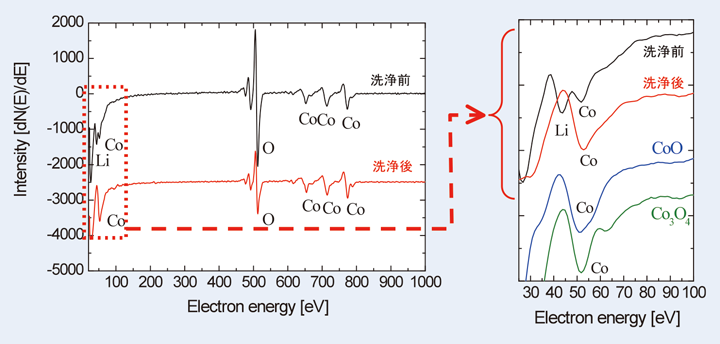
【Fig. 1 LiCoO2表面のLiピーク強度の違い(超音波洗浄前と洗浄後)】
2.2 オージェ分析時の粉末試料のための前処理法と注意点
粉末試料に対して表面分析を行う場合の、試料前処理法や分析のコツについては、2009年EPMA・表面分析ユーザーズミーティング資料[1]で説明した。ここでは特にLi含有粒子に対して有用である「カーボン試料台に散布する方法」を紹介する。
カーボン試料台とは、Fig.2に示す日本電子(株)で販売している「水平試料載台」(Parts No. : 600154386)のことで、その大きさは直径10mm、高さ5mmである。この試料台を入手して、その表面を粒度1000番程度の研磨紙で研磨し、ある程度平滑な表面とした後、その表面上に精密ドライバーなどの先端の尖った工具で少し浅い引っかき溝を作る。このように処理したカーボン試料台の上に分析目的である粉末試料を散布した後、薬包紙などの紙で粉末試料を散布した表面を軽く擦って、粉末を溝に擦り込み、擦り込むことのできなかった粉末は除去する。さらにブロアー等で、カーボン試料台表面に残っている余分な粉末を除去する。
このように試料前処理を完了したカーボン試料台を、 Fig.3に示すようにAES用の標準試料ホルダーの内部にあるスプリングを外してセットすれば、AESのための準備は終了である。
この試料前処理を行うと、カーボン試料台の上に様々な形態の粒子が存在することになる。例えば、Fig.3の右図の二次電子像に示すように、先ほど作った溝のところには、粉末が充填されて密に粒子が詰まった大きな表面が再現できている。また、1000番の研磨紙で研磨した表面には微細な凹凸や研磨痕があり、単一状態の粒子やクラスターを形成している粒子群が凹凸や研磨痕中に存在している。このように、一度の前処理でカーボン試料台の上には、様々な形態で粒子が存在しているので、この中から単一粒子を選び出して分析したり、密に詰まっている領域で平均的な組成を分析したりするなど、一つの試料でいくつかの状態の分析が可能となる。

【Fig.2 カーボン試料台への粉末試料のセッティング。】

【Fig.3 標準試料ホルダーにカーボン試料台をセットした状態と表面二次電子像。】
2.3 XPS分析時の粉末試料のための前処理法と注意点
XPSにより絶縁性の粉末試料を測定する場合には、試料が絶縁物であることにより帯電の影響を受けること、試料ホルダーへの固定が難しいため飛散しやすいことなどの問題がある。他の固定方法との比較については2006年EPMA・表面分析ユーザーズミーティング資料[2]に報告した。XPS用試料成型としての最適な方法は錠剤成型器を用いてペレットに成型することである(Fig.4)。ペレット成型を行うためには比較的多量の粉末試料を必要とするが、この方法で成型された試料表面はフラットであるため、 XPSにおいて感度が高く、帯電の影響も抑えやすい。

【Fig.4 ペレットに成型された粉末試料。】
3.表面分析におけるLiの検出と定量
3.1 Liに関する検出と感度
AESにおいては、Liに関する感度は思いのほか低くはなく、点分析はもちろんのことマッピングも短時間で行うことができる。そこで、Liの検出感度についてAESとXPSとの比較を行った。Fig.5の左図に示すように、AESによってC(カーボン)とLiを同じ条件で測定した場合の標準スペクトルのピーク強度を比較すると、Cに比べてLiの方が4倍程度高いことがわかる。一方、XPSの場合も本来であれば純物質同士のピーク強度を直接比較すべきところであるが、Liの標準スペクトルの取得が困難であるため、これができない。そこでAlKα(1486.6 eV)のX線で励起する際のイオン化断面積を、CとLiの感度として比較した(Fig.5 右)。これによるとAESの場合とは逆になっており、LiはCに比べて、1/18程度のピークであることがわかる。つまり、Cの強度を基準にLiの強度を比較すると、約72倍の違いがあり、 AESの方が高い感度を有している。
しかし、一般的にはXPSの方がLiを検出しやすいという印象があり、実際、緒言で述べたようにLiの表面分析は、多くの場合XPSで行われている。これはLiのオージェ電子とLiの光電子の運動エネルギーの違いによる脱出深さの差が大きく関与している。Fig.6に固体試料中の電子の平均自由行程のエネルギー依存性を示す。Li KVVのオージェ電子は、Li 1s軌道の結合エネルギーの大きさを反映して約50eVの運動エネルギーしか有していない。これに対し、AlKα線を用いたXPSでのLiピークは一次X線のエネルギーを反映した1400eV以上の高い運動エネルギーを持っている。このため、平均自由行程が大きく異なっている。電子の脱出深さを平均自由行程の約3倍程度として見積もると、Li KVVオージェ電子の脱出深さは約2nm弱となり、これはXPSの光電子のそれの1/3 程度と著しく浅い。このため、わずかなコンタミネーションが付着しても、その強度は著しく低下し、検出できなくなる可能性が高いことがわかる。つまり、Liに関するAESを行う場合には、コンタミネーションを除くための試料前処理方法も非常に重要である。
次に実際にXPSを用いて分析を行ったLi化合物のスペクトルを示す [3]。まず金属Liの測定を行った結果を示す。金属Liは通常酸化を避けるため、パラフィン油中に浸して保存される。ただしLiは室温でも水と反応し、水酸化リチウムとなり、その後大気中の二酸化炭素を吸収して炭酸リチウムへと変化する。これにより保存していたLiを大気中に取り出すとLiの周辺は白く変色した状態である(Fig.7)。この状態のLiをカミソリで切ることにより金属光沢を持った面を露出させることができる。しかし大気中では金属光沢を示していた部分も数十秒のうちに黒色、そしてその後白色へと変化していく。
炭酸リチウムに変化した白色の部分は長時間イオンスパッタリングを行うことによって表面をエッチングしても酸素や炭素はなくならず、金属Liのスペクトルを取得することはできなかった。Fig.8にXPSで測定した炭酸リチウム部分のスペクトルを示す。
Liは大気中の水分と反応して変化するため、Liの塊をドライ窒素を充満させたグローブバッグ内で切断し金属光沢を保った状態のままXPSを行うと、金属Liと酸化リチウムに対応するスペクトルが得られる。このスペクトルとFig.8に示した炭酸リチウム、さらにリン酸リチウムの3種類の化合物からのLi1sスペクトルをFig.9に、それらの化合物ごとのピーク位置をTable 1に示す。このようにXPSを用いることにより、Liのスペクトルが明確に観察されるとともに、 Li自体の化学状態の変化をもとらえることができることがわかる。
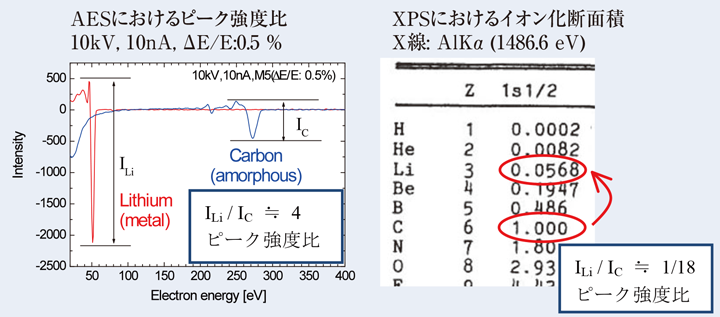
【Fig.5 AESとXPSのLi感度の比較。】
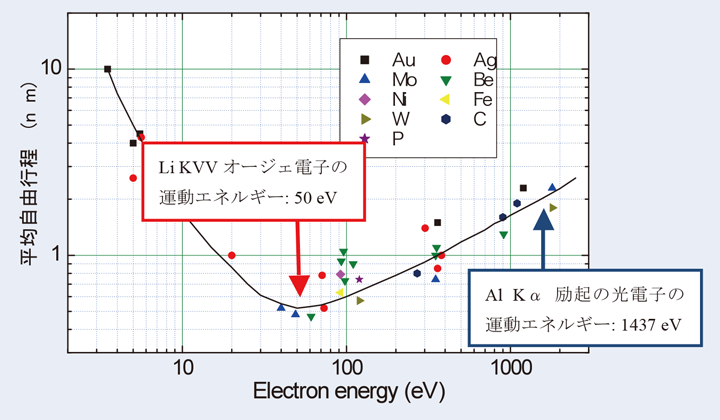
【Fig.6 固体試料中の電子の平均自由行程。】

【Fig.7 Liメタルの写真(左:切断前、右:切断直後)。】
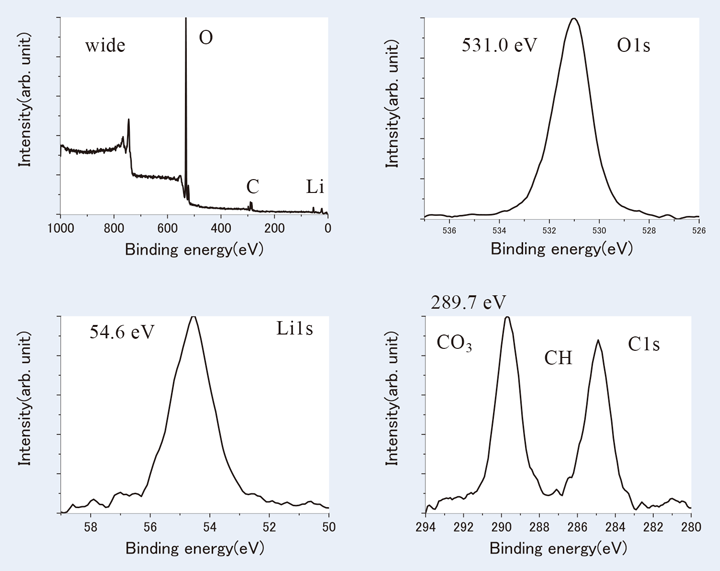
【Fig.8 Li白色部分のXPS測定結果。】
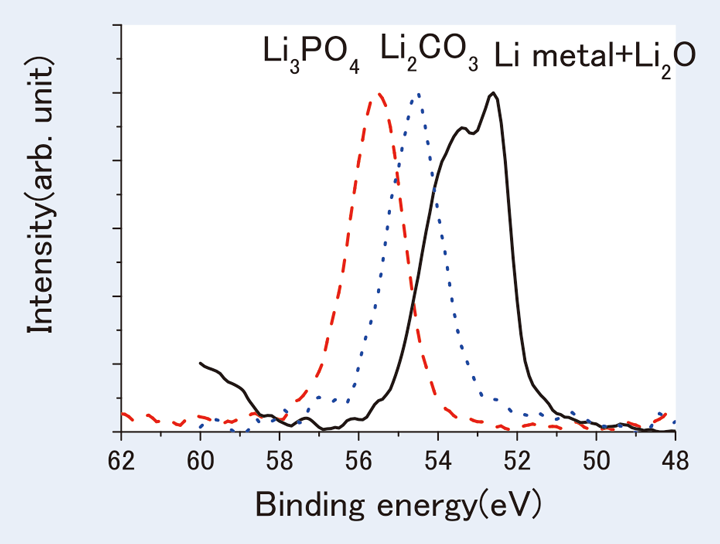
【Fig.9 各化学結合状態のLi1sピーク形状。】
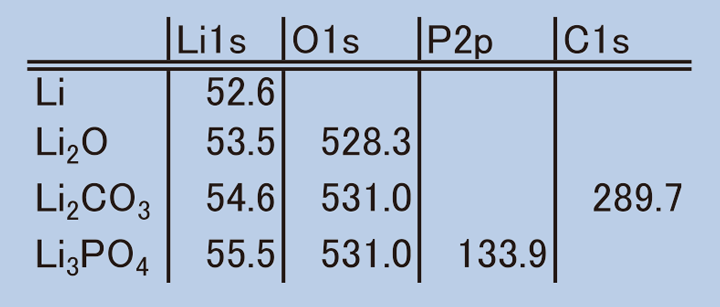
【Table 1 Li関連物質のピーク位置。】
3.2 Liの定量 (相対感度因子法と絶対強度定量法)
前節までに試料前処理方法とLiを検出する際の注意点について述べた。上記の点に注意を払えば、AESにある程度習熟したオペレータはXPSのみならず、AESによってLiの分析はさほど困難なく可能である。しかし、その次の問題点であるLiの定量分析と定量精度については、現在のところ確立した方法がない。ここでは、Liの定量分析法として相対感度因子法と絶対強度定量法について述べる。
Liは1sと2sの2つの電子軌道にしか電子を有しておらず、このうち2s軌道は結合している相手元素との共有結合軌道となるので、オージェスペクトルは結合状態によってピーク位置とピーク形状が大きく変化する。また、50eV付近のLiのKVVピークは二次電子の大きなバックグラウンドの上に存在し、その50eV付近は他の元素の価電子ピークがオーバーラップするために、従来のAESの定量分析法として用いられている相対感度因子法では、正しく原子濃度を見積もることは非常に困難である。
Fig.10に相対感度因子法に用いるAgとCuの強度を示した。AESにおける相対感度因子法では、実測された微分スペクトルの各元素の山谷部のピーク強度 (Fig.10)に、予め求められている純物質間での相対ピーク強度比(相対感度因子)をかけて感度の補正をした後、それらの値を合計して100 %になるように規格化したものを原子濃度比として求めている。この方法は非常に簡便な方法で、相対感度因子さえ求まっておれば、実測したオージェスペクトル1本から各元素の微分ピーク強度を求めて単純計算をするだけで、定量分析値を得ることができる。相対ピーク強度(相対感度)を考慮して規格化するということでは、基本的にはXPSにおける定量計算も同じ考え方であり、表面分析では広く認知された一般的な定量分析の方法である。しかしAESの場合、XPSの場合と異なり、あくまで微分ピークの山谷部のピーク強度 (Fig.10)を基準とするため、微分ピーク強度はピーク形状の変化に著しく敏感で、結合状態変化によるオージェピークのブロード化や、他の元素ピークのオーバーラップによるピーク形状の変化は、定量結果の誤差を大きくする要因となる。これらの要因は、Liを定量する場合は非常に深刻な問題で、原子濃度が5%以下となるような低濃度のLiピークの場合などでは、もはや独立したピークとして山谷部を検出することはできず、波形分離計算の結果で初めて他の元素のピークの中にLiが含まれていることがわかる場合も少なくない。
そこで、Liの定量分析を行う場合には、相対感度因子法ではなく、ここでは絶対強度定量法を用いて行うことを提案する。手順は、次のステップ(1)~(4)の通りである。
(1) 試料前処理に注意しながら、オージェスペクトルを測定
(2) 実測した微分スペクトルに対して、標準スペクトルを使った波形分離計算による各元素スペクトルへの分離
(3) 各結合状態の元素スペクトル強度と、それらと同じ測定条件下で測定した場合の標準スペクトル強度との強度比を用いた原子濃度への変換
(4) 検出された原子濃度の総和による定量精度の検討
ここでステップ(1)で測定するオージェスペクトルのエネルギー分解能は、必ずしも高いエネルギー分解能を選択する必要はない。一般的なエネルギー分解能0.5%でも、同じエネルギー分解能で測定された標準スペクトルを有していれば定量計算には差支えない。もちろん、高エネルギー分解能で測定すれば、結合状態や原子価に応じた標準スペクトルを使った波形分離計算が可能となり、結合状態別の原子濃度まで定量が可能となる[4]。解析目的にあったエネルギー分解能で測定し、電子線やArイオンによるダメージに気を付けながら測定を行うことが重要である。ステップ(2)では、標準スペクトルを用いて、最小二乗法により残差(誤差)が最も小さくなるように計算を行う。具体的な計算方法については、ここでは割愛するので、参考文献 [5]の「AESのピーク分離解析とその応用」を参照していただきたい。ここで重要となるのは、波形分離を行う場合に、必ずスペクトルを微分して、簡易的にバックグラウンドを除去することである。オージェスペクトルの場合、オージェ電子そのものが二次電子の一種なので、微分する前のN(E)スペクトルに含まれる二次電子のバックグラウンド成分とオージェスペクトル部分を明確に区別することができない。微分することで、簡易的に二次電子のバックグラウンドを除去することで計算精度が向上し、ピーク形状の差異に特化した波形分離計算が可能となる。
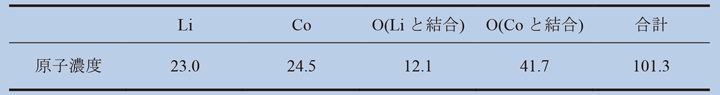
ステップ(3)では、ステップ(2)で得られた各元素のスペクトルの強度と、標準試料を同じ条件で測定した場合のスペクトル強度とを比較して、絶対強度定量計算を行うことになる。ここでFig.1で紹介したLiCoO2粒子について測定したスペクトルに対して、絶対強度定量を行う手順をFig.11を使って説明する。なお、ここでは複合酸化物であるLiCoO2をLiO2とCo3O4の混合酸化物であると仮定している。
Fig.11に示したスペクトルは、LiCoO2 粒子について照射条件(10kV,10nA)、エネルギー分解能0.5%で測定したものである。このエネルギー分解能であると、Co2+ やCo3+ の価数による結合状態の違いをスペクトルで区別することは難しく、Coのスペクトルはほぼ同じ形状をしているので、絶対強度定量法に用いる標準スペクトルはCoOでもCo3O4でも問題はない。ここでは酸化コバルト(Co3O4)と酸化リチウム(Li2O)の標準スペクトルを使って定量計算を行った。
Liが検出される30~60eV付近のピークにはCoのピークも検出されるので、酸化リチウムと酸化コバルトの標準スペクトルを使って波形分離すると、Li2O成分が抽出される。今回の場合の、その強度は1078countsで、同じ測定条件で測定した場合のLi2Oの標準スペクトルの強度は3127countsである。これは波形分離されたLi2O成分の絶対強度が3127counts となれば、標準試料のLi原子濃度と同じ2/3=66.66 …%になることを示している。そこで、絶対強度比から測定されたLi濃度は約23.0%であることがわかる。同様に、Coが検出される600~850eV範囲は、この場合Coの3つのピークしか存在しないので、Co3O4標準スペクトルとの絶対強度を比較して、Coの原子濃度は約24.5%であると計算される。
最後に、470~530eV付近に存在する酸素のピークはLiに結合する酸素ピークとCoに結合する酸素ピークが重なって存在しているが、ピーク位置がそれぞれ異なるので各標準スペクトルを使って波形分離計算を行うと、2つの成分に分離した絶対強度を得ることができる。その結果、得られた絶対強度比から、それぞれの成分の原子濃度を計算すると、Liと結合する酸素は約12.1%であり、Coと結合する酸素は41.7%であることが求められる。
それぞれの絶対強度比から求めた各原子濃度と合計の原子濃度をTable 2に示した。原子濃度の合計値を見ると、ほぼ100% でその差は1.3% 程度になっている。相対感度因子法では必ず規格化されるので、定量計算結果の誤差に対する議論が難しくなるが、絶対強度定量法ではこの原子濃度の合計値を見れば、誤差をおよそ見積もることができる。また、各原子濃度比も重要なポイントである。LiとLiに結合している酸素の比は約2:1で酸化リチウムを形成していると推定でき、Li:Co:O(合計原子濃度)は1:1:2と、ほぼLiCoO2を形成していることが明らかとなった。
このようにLiのピークのように、他の元素のピークがオーバーラップしても、波形分離後のスペクトルに対して、絶対強度定量法を用いれば、Liの原子濃度を求めることが可能である。
一方でXPSの場合には、3.1節で示したようにLiのスペクトルの化学結合状態による変化は、主にスペクトルのシフトだけであること、また相対感度因子法による定量計算ではスペクトルの面積強度を用いることからLiの原子濃度を計算するときにも相対感度因子法によるもので比較的精度のよい結果が得られる。ただし、XPSの場合に気をつけるべき点は試料の不均一さである[6]。粉末の試料の場合にも試料自体が不均一であることが十分にあり得る。
Fig.12の反射電子組成像よりペレット成型した粉末試料はおよそ100μm程度の不均一さを持つことがわかるが、通常XPSによる試料の観察は光学顕微鏡などでなされるため、光学顕微鏡で観察される以下の不均一さを判断することは難しい。このような場合には分析径を十分に広げることによりその影響を軽減することが可能となる。Fig.13はFig.12の試料を分析径100μmφ、および3mmφとして定量分析を行った結果である。Fig.13より分析径100μmφの場合には分析位置により特にLiの量に大きな違いが存在していることが認められる。一方で分析を3mmφとした場合にはそのような定量値の違いは認められない。このようにXPSの場合には特に試料の不均一さを意識したうえで分析を進める必要がある。
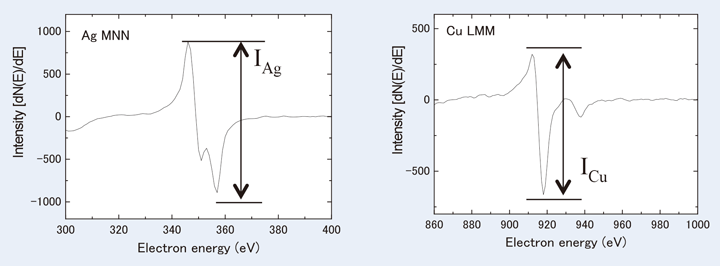
【Fig.10 相対感度因子法における強度の定義。】

【Fig.11 絶対強度定量法(Fig. 1のLiCoO2スペクトルの定量計算例)。】

【Fig.12 ペレット成型したLiイオン電池用粉末材料とその反射電子組成像。】
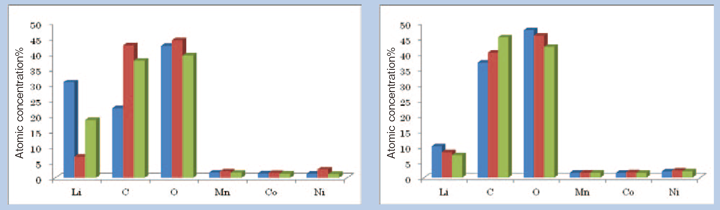
【Fig.13 左:任意の3点を分析径100 μmφとして測定した定量結果。右:任意の3点を分析径3 mmφとして測定した定量結果。】
【Table 2 絶対強度定量法で求めたLiCoO2原子濃度(%)。】
3.3 Li電池用粉末材料のオージェ分析と他の分析法との比較
ここでは実際にLiイオン電池用粉末試料のAESによる解析事例を紹介し、他の分析法による結果と比較する。ここで扱う試料は、Fig.14に示すMn,Co,Niが1:1:1の割合で含有されているNMC系粉末試料(以下NMC試料)で、これを適切な試料前処理を行って分析した。
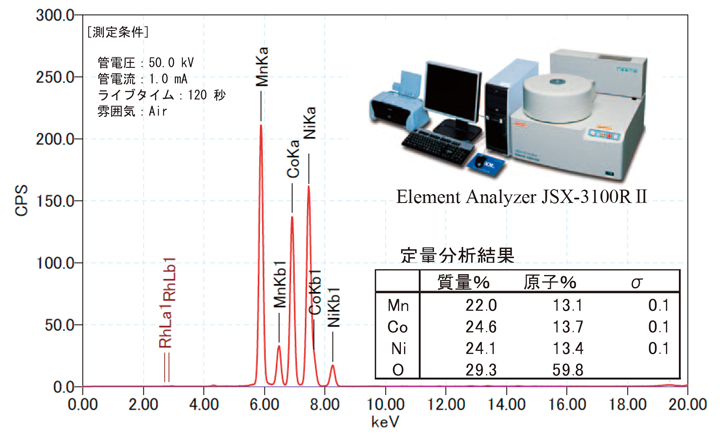
まずAESで分析する前に、平均組成が指示通りの比率Mn:Co:Ni=1:1:1の比率になっているか否かをエネルギー分散型蛍光X線分析(XRF)装置 [Element Analyzer JSX- 3100RⅡ]を用いて調べた。一般的にLiイオン電池用粉末材料の平均組成を調べるためには、主に湿式分析によるICP-MS(誘導結合プラズマ質量分析計)、ICP-AES(誘導結合プラズマ発光分光)などの方法が用いられることが多い。しかし、湿式分析は定量精度が高いという大きな長所もあるが、試料を酸・アルカリ溶液に全溶解させて調べる必要があり、溶液調整に時間が必要となるため、多くのサンプル数をこなすことが難しいという欠点もある。そこで、Liイオン電池用粉末試料に対してLiの含有量を除いたMn,Co,Niの比が正しいか否かに関しては、一次スクリーニング手段として、わずか数分で精度の高い定量結果が得られるXRF法が用いられている。Fig.15にXRF法で観測されたスペクトルおよびこのスペクトルを用いて得られた分析結果を示す。この結果を見ると、XRFの分析結果ではMn,Co,Niはどれも原子濃度約13%で、ほぼ1:1:1の割合で含有されていることがわかる。
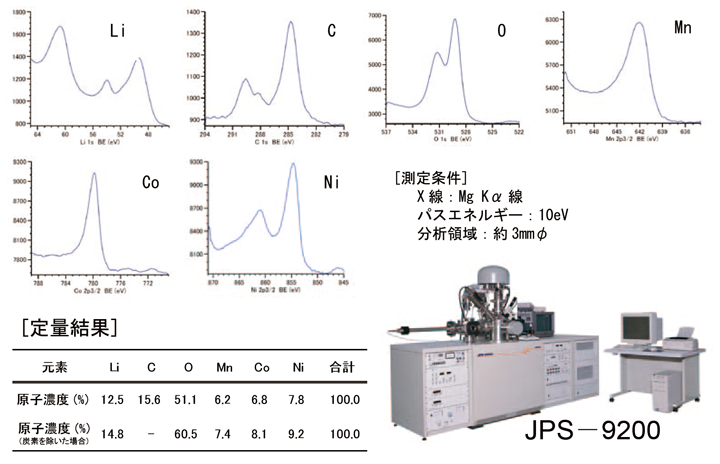
次に、粉末粒子をペレットにして、粒子表面に平均的に存在するLi濃度を調べるため、XPSによりJPS-9200を使って、NMC試料の最表面分析を行った。Fig.16にその結果を示す。XRFの結果と比べると、ややNiの濃度が他のMn,Coの濃度よりも高い値を示しており、表面には14.8%ものLiが存在していることがわかる。
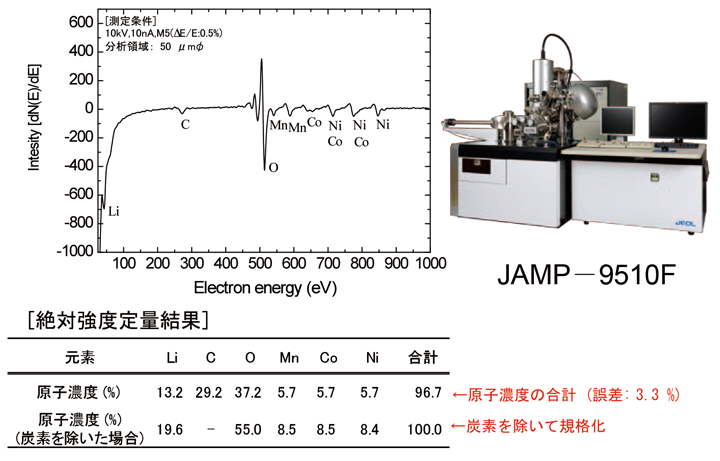
続いて、AESを用いて表面における平均組成を分析するために、同じ試料で50μmφの領域での平均スペクトルを測定した(Fig. 17)。測定されたスペクトルに対して、3.2の節で述べた方法で絶対強度定量法による定量分析を行い、その結果もFig.17に示す。絶対定量分析結果では、各元素の定量値(原子濃度)はそれぞれのピーク強度を標準スペクトルのピーク強度と比較して求めるため、規格化は行わない。今回の結果は総原子濃度の合計値が96.7%となり、誤差は約3.3% であることがわかる。参考までに、コンタミネーション由来と考えられる炭素を除いて規格化した定量値で、従来の相対感度因子法と絶対強度定量法と比較した結果をTable 3に示す。相対感度因子法で計算すると、Liピークは他のMn,Co,NiなどのMVVピークがオーバーラップするため、その濃度が絶対強度定量法の濃度よりも大きな値となっていることがわかる。
XPS(Fig.16)とAES(Fig.17)との定量結果を比較すると、炭素濃度の違いが大きいことが目立つが、これは恐らくXPSとAESの分析領域の違いによるものだと考えられる。平均的な元素組成を分析する場合、広い領域を分析することが可能であるXPSの方が有利であり、コンタミネーションが局在していても影響を受けにくい。一方、AESは元来局所分析を目的としており、アナライザーのレンズ系の都合上、分析範囲は最大数100μm四方が限界で、今回無作為に平均的と思われる場所を測定したが、偶然にも有機物の多い領域を分析したために、炭素濃度が高かったと考えられる。そこで、炭素を除いた元素で規格化した定量値で比べてみると、AESとXPSは近い値になり、Liについては5%程度の差に収まっている。このように、AESを使ってLiの定量を行う場合には、絶対強度定量を用いた方がより精度の高い分析が可能となるので、活用していただきたい。
次に、2.2節で紹介した様に、カーボン試料台上にNMC試料を分散させて二次電子像で観察した結果をFig.18に示す。
Fig.18を見ると、大きさの異なる粒子が数多く存在しており、中には有機物と思われる異物も確認できる。ここでは、簡便のために粒子径の大きいものをpoint 1、粒子径の小さなものをpoint 2、異物をpoint 3としてこれらの点について点分析を行った。生のスペクトルとこれらのスペクトルから求めた分析結果をFig.18に示す。その結果、point 1とpoint 2とではMn, Co, Niの原子濃度比が異なっており、これらはLiの濃度も大きく異なる粒子であることがわかった。また、point 3ではLiは全く検出されず、これは単なる異物だと思われる。一般にLiイオン電池用粉末試料中には、このような異物が多く混入しており、分析領域によってはFig.17で得られた結果のように炭素濃度の高い定量値が得られているが、この結果はこのような観察で説明できる。このように、Liイオン電池用粉末試料の場合には、広い領域を平均的に分析するXPS等の結果と、局所的に分析するAESの結果とは、異なることがある。そのため、試料をより正確に解析するためには、どちらか一方のデータだけでは不十分で、平均的なデータと局所領域のデータを取得し、これらから試料の分析結果を総合的に判断する必要がある。
続いて、このNMC試料断面をクロスセクション・ポリッシャー(CP)で作製し、断面に垂直な方向から分析を行った(Fig.19)。AES用のLiイオン電池用粉末試料の断面を作製する場合には、いくつか注意するべき点がある。第一はLiの拡散を防ぐため、粒子の固定はカーボン試料台を用い樹脂等の使用は避けること、第二は粒子内の試料の状態を保持するため、断面加工操作は粒子内の元素と化学反応を起こさない不活性ガスイオンで加工するCPやイオンスライサーなどの断面加工装置を用いること、第三は断面加工した表面を変質させないために、大気に曝すことなく試料を保持・搬送するトランスファー・ベッセルなどを活用することである。特に、AESは表面からわずか数nmといった領域で、元素分析や結合状態分析を行う装置であるので、細心の注意が必要である。
Fig.18を4倍に拡大したFig.19の二次電子像を見ると、試料の粒子は内部に空胞を持っており、ポーラス状の構造を持っていることがわかる。Liイオン電池用粉末試料の中には、原材料粒子を混合して焼成する過程で、Fig.19に示すような粒子内部に空胞を有していることがある。CPは不活性ガスを用いたドライプロセスであって、このようなポーラス状の構造をもつ粒子でも穴の形状を保ったまま断面加工できるため、Liイオン電池用粉末試料の分析に適している。この粒子断面上の比較的構造が密な図中に丸印で囲った部分について、オージェスペクトルを測定し、絶対強度定量分析を行った。その結果をFig.19の表に示す。Mn,Co,Niはおよそ1:1:1の割合で存在しており、炭素を除いた原子濃度に換算すると、その濃度は約13~14%になる。この値はFig.15のXRFの結果に近い値であり、粒子そのものの原子濃度を示していると考えられる。
次に、Fig.20のSEIに示すように、Fig.19に示すものと同じ断面試料を使って、同じ丸印で囲った場所をSEM-EDSで分析した結果をFig.20の表に示す。AESの結果と比べてみると、Mn,Co,Niの原子濃度では、わずか2%程度しか異なっておらず、ほぼ同じ値を示していることがわかる。
ここまでの結果をまとめると、Table 4のようになる。表面分析においても、バルク分析においても、AESによる絶対強度定量分析の結果は、他の分析法の結果と比較しても、 Mn,Co,Niの原子濃度の差違は約2%程度以内に留まっており、Liにおける定量値の誤差も数%程度だと推定される。このように、絶対強度定量法は、Liの定量においても、妥当な精度を有しており、AESを用いればLiイオン電池用粉末試料分析において、局所分析から平均領域分析まで幅広く活用できると考えられる。

【Fig.14 実験に用いたLiイオン電池用NMC系試料(Mn, Co, Ni = 1:1:1)。】
【Fig.15 蛍光X線分光分析(XRF)によるNMC試料の定量分析結果。】
【Fig.16 X線光電子分光法(XPS)によるNMC試料の定量分析結果。】
【Fig.17 AESによるNMC試料の定量分析結果(絶対強度定量法)。】
![Fig.18 Liイオン電池用NMC系粉末試料[10kV,10nA,観察倍率:700倍]](./product_file/file/n46-06-fig18.png)
【Fig.18 Liイオン電池用NMC系粉末試料[10kV,10nA,観察倍率:700倍]。】
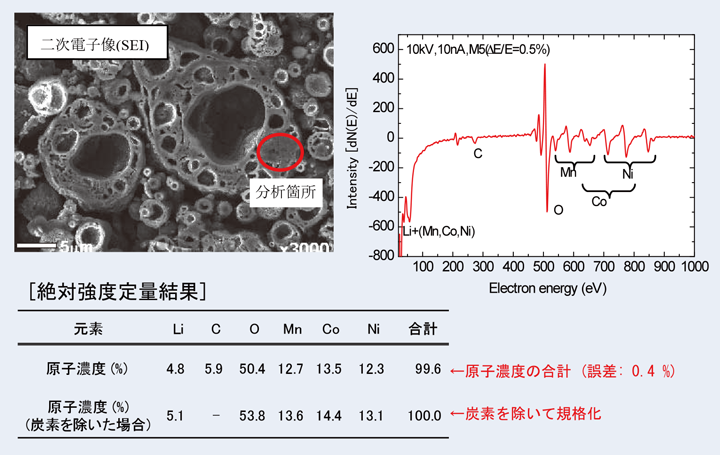
【Fig.19 NMC粉末断面で測定したAESスペクトルと絶対強度定量結果。】
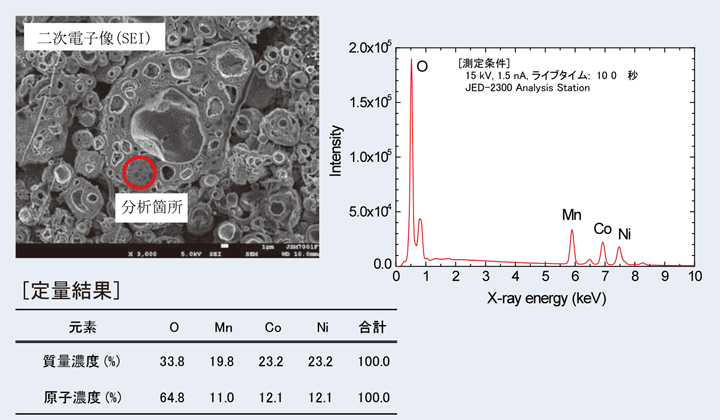
【Fig.20 SEM-EDSによるNMC試料の定量分析結果。】
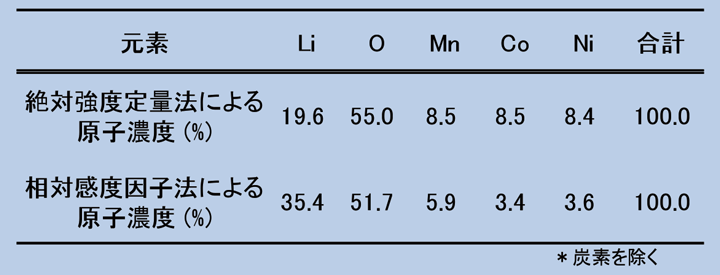
【Table 3 NMC試料に関するAESにおける絶対強度定量法と相対感度因子法との定量値の比較(炭素を除いて規格化)。】

【Table 4 各種分析装置を用いたLiイオン電池用粉末試料の定量結果の比較。】
4.結言
Liイオン電池は、今日世界中の研究者によって精力的な開発が進められており、数多くのレポートも発表されている。その中で、電池としての電気特性向上の開発の基礎となる電池の中の局所領域におけるLiの挙動およびLiの状態に応じた分布に関する知見が強く求められている。AESやXPSはまさにそのようなニーズに応えることができる数少ない手法であり、Liの検出・定量が可能であるばかりでなくAESではナノ領域におけるマッピングまで可能な装置として市販されている。しかし、特にAESではLiの分析は困難だという偏見があって、現状では残念ながらこの目的のためにあまり活用されていない。
今回、ここで紹介したようにXPSを用いたLiの分析だけでなく、AESを用いることでもLiの高感度の分析が可能であり、絶対強度定量法を用いれば他の分析手法と比較しても、同じ程度の信頼性のおける定量値を得ることもできる。AESやXPSを 十分に活用できれ ば、TEM,SEM,EPMA,XRFなどの直接Liを測定できない装置のデータを補完することができ、これらのデータを総合的に判断することで、今まで明らかにされていなかったLiの分布や挙動を明らかにできるものと期待できる。
参考文献
[ 1 ] 堤建一 :「Li電池・太陽電池開発におけるAES化学状態分析の応用」, 2009 EPMA・表面分析ユーザーズミーティング資料, AP100 (2009).
[ 2 ] 島政英 :「絶縁性粉末試料のサンプリングとXPS分析テクニック」, 2006 EPMA・表面分析ユーザーズミーティング資料, XP41 (2006).
[ 3 ] 島政英 :「XPSによるLiイオン電池関連材料の化学結合状態分析」, 2010 EPMA・表面分析ユーザーズミーティング資料, XP45 (2010).
[ 4 ] 堤建一,島政英,田中章泰,田澤豊彦: 「高エネルギー分解能AESによるSn, SnO, SnO2の化学状態定量分析」,表面科学 Vol. 33, No. 8, pp. 431-436, (2012).
[ 5 ] 堤建一 :「AESのピーク分離解析とその応用」, 2001 EPMA・表面分析ユーザーズミーティング資料, AP92(2001).
[ 6 ] 島政英 :「XPSにおける広領域分析の有効性-微小領域分析の落とし穴-」, 2012 EPMA・表面分析ユーザーズミーティング資料, AP92 (2012).