固体NMR・X線回折・電子回折の複合アプローチによる低分子有機物の構造解析法
日本電子news Vol.50 No.5
西山 裕介1, 2
1 株式会社JEOL RESONANCE 2 理化学研究所CLST-JEOL連携センター
本稿では、われわれの研究室で近年開発した手法および応用研究の中から、低分子医薬品の構造決定を行うための複合アプローチを取り上げたい。低分子医薬品では、安定性・薬効・安全性のみならず知財の確保の面からも1)結晶形の同定、2)塩/共結晶の決定といった原子レベルの構造解析が必要になる。TEM(transmission electron microscope)における高感度カメラの出現や、固体NMR(nuclear magnetic resonance)における70 kHzを超える高速MAS(magic angle sample spinning)技術の進歩はこれらの問題を解決する上で重要な道を拓きつつある。1)結晶形の同定には、粉末X線回折および固体13C NMR測定を用いた複合アプローチは幅広く用いられているが、決して万能というわけではない。そこで、電子回折および固体1H NMRによる相補的な手法を提案する。これにより、製剤中に含まれる微小結晶からの結晶形の同定や、水素位置の決定などが可能になる。2)塩/共結晶では、分子間相互作用(イオン結合もしくは水素結合)の理解のため水素位置の決定が必須である。X線回折法は、炭素・酸素・窒素といった低分子医薬品の骨格構造を明瞭に決定できるが、水素位置の決定精度は高くない。そこで、X線回折法による骨格構造の決定と固体1H NMR法による水素位置の精密決定を組み合わせることにより、塩/共結晶の問題に明瞭な解を与えた。
はじめに
近年バイオ医薬品が売り上げ上位を占めるようになりつつあるが、経口による服用が不可欠である生活習慣病をはじめとする日常の治療において、低分子医薬品が重要な位置を取り続けることは明白である。多くの低分子医薬品のAPI(active pharmaceutical ingredient)は良好な結晶性を示し、錠剤・顆粒剤・散剤など賦形剤を加えた微結晶粉末で提供されている。これらの医薬品の効能は結晶形により大きく影響を受けるため、結晶多形の区別・塩/共結晶問題といった原子レベルの構造解析法が研究開発から品質管理にいたるまで要求され、日常的に測定が繰り返されている。特に結晶形の同定の目的には、粉末X線回折法および固体NMR(nuclear magnetic resonance)測定が相補的に幅広く用いられている。粉末X線回折法および13C CPMAS(cross-polarization magic-angle-spinning)固体NMR法を用いることにより、ピーク位置から結晶形の区別ができる。粉末X線回折は明確に結晶形を区別できるが信号の重なりに弱いという問題を抱えている一方で、13C CPMASでは賦形剤とAPIの信号が異なる位置に現れ、信号の重なり合いの問題が深刻ではないが結晶形の分離能は粉末X線にやや劣る。このように医薬品研究において固体NMR法と粉末X線回折による複合的アプローチは、従来からお互いの不足能力を効率よく補完しあう手法として幅広く用いられてきた。しかしながら、これらのアプローチも万能ではなく、1) 水素位置の精密決定ができない 2) 必ずしも明瞭に結晶形が区別できるわけではない、3) 試料によってはスループットが低い、といった問題を抱えている。
ここで、われわれは複合的アプローチを改めて見直し、スループットと情報量を両立する測定法を検討する。まず、われわれが注目した各手法の利点と問題点についてまとめておきたい。単結晶X線回折は原子レベルの構造情報を与える非常に有用な手法である。その一方で、一辺10 ~ 100 μm程度の非常に大きな単結晶試料が必要になり、微結晶としてAPIが含まれている製剤の解析を不可能としている。さらに、水素位置の精密決定ができないことも問題である。粉末X線回折は、結晶形の同定に有用であるものの信号の重なりに大きく影響され製剤の解析にしばしば問題をきたす。13C CPMAS固体NMR法は、ピーク位置がコンホメーションに敏感である一方で、パッキングの違いはあまり反映されない。また、全体構造を直接決定できない点も問題である。本稿では、これらの問題を解決する相補的手法として電子回折法および1H固体NMR法の活用を提案する。電子回折は透過型電子顕微鏡(TEM: transmission electron microscope)の測定モードのひとつであり、単結晶X線回折のように非常に大きな結晶を必要としない。これは電子がX線と比べて104 ~105強い相互作用を示すことが理由であり、nm ~サブμmサイズの非常に微細な単結晶試料からも十分な回折強度を得ることができる。幸い製剤中に含まれるAPIの結晶サイズは、単結晶電子回折を得るのに十分大きい電子回折で単結晶回折パターンを測定できる。このように電子回折は非常に大きなポテンシャルを持ちながらもサンプルが電子損傷を受ける問題があり、有機物への適用は限定的であった。しかしながら、近年の高感度検出カメラの開発やクライオホルダーによる熱損傷の低減により、APIの電子回折測定が現実的になった。1H固体NMR法は、回折法全般の抱える水素原子の位置決定ができない問題を補う。幸い、1H核は安定同位体の原子核のうちでもっとも核磁気回転比が大きく、他の核と非常に強く相互作用を示し、また、99%以上の天然存在比とあいまって非常に強い信号強度を示す。そのため、原理的には多くの核間相互作用を高感度で測定できる潜在能力を持っている。その一方で、固体試料中では強すぎる核間相互作用のために現実的には分解能が非常に低く、固体NMRにおいて1H測定は構造解析にほとんど用いられてこなかった。しかしながら、近年の高速MAS技術の進歩により[1-3]、70 ~ 100 kHzといった従来よりも一桁速い試料回転が実現し、APIを含む固体試料の1H NMRの高分解能測定が実現した。
上記の状況を踏まえ、本稿ではX線回折・電子回折・固体NMR法を有機的に結合させることにより、結晶多形・塩/共結晶問題に解を与える複合的アプローチを提案する。以下の章では、まず結晶多形への電子回折と固体NMRの適用を議論する。またその中で開発した新規手法を紹介する。次に塩/共結晶問題に明瞭な解を与えるX線回折と固体NMRの複合アプローチおよび、スループットの改善のために新規開発した手法を紹介する。なお本手法は、従来の手法を置き換えることを意図したものではなく、従来法と相補的に用いることによりスループットと結果の最適化を目指したものである。
結晶多形[4]
医薬品の溶解度・安定度といった物性は、結晶形に非常に大きく左右されるため、結晶形の同定とコントロールは医薬品にとって必須の技術である。幅広く用いられている粉末X線回折および13C CPMAS測定は、先に議論したとおり常に結晶形を明瞭に区別できるわけではない。たとえば、L-ヒスチジン塩酸塩一水和物とL-ヒスチジンの13C CPMASスペクトルはコンホメーションの違いを反映して明瞭に区別できるのに対して(Fig. 1)、L-ヒスチジンの二つの異なる安定結晶形であるorthorhombic形とmonoclinic形の13C CPMASスペクトルは、非常に似通っておりお互い重なり合い区別ができない(Fig. 1: b, c)。これはこの二つの結晶形では、分子のコンホメーションがほとんど同じであり、結晶形の違いは分子間パッキングの違いから生じていることが原因である。そこで、これらの従来法を補完する手法として、本稿では、電子回折および高速MASのもとでの1H NMRの活用を提案する。
Fig.1
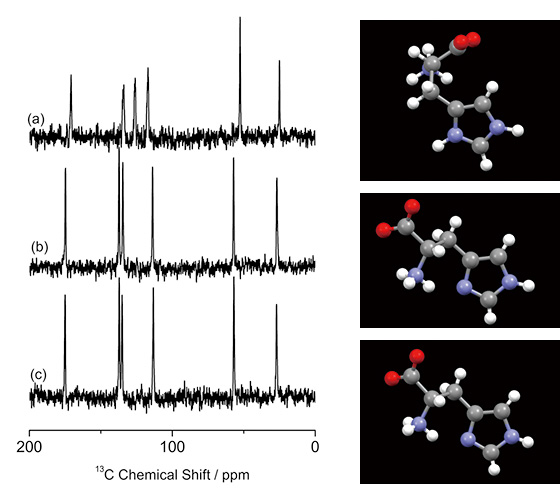
(a)L-ヒスチジン塩酸塩一水和物、(b)L-ヒスチジン(orthorhombic)、(c)L-ヒスチジン(monoclinic)の13C CPMASスペクトルおよびコンホメーション。(参考文献4より引用)
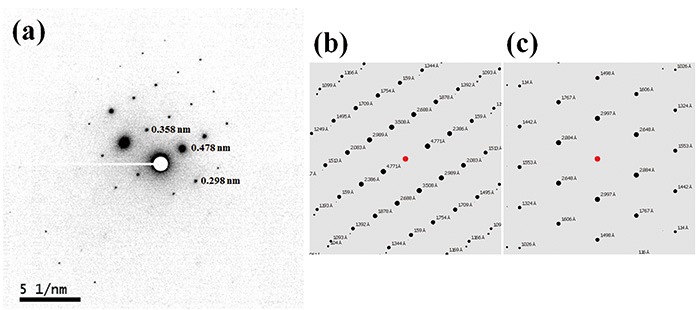
Fig. 2に常温で測定したL-ヒスチジンン塩酸塩一水和物のTEM像および電子回折パターンを示す。先に述べたように電子回折により、製剤に含まれるサブμmの非常に小さな単結晶からの回折パターンが容易に得られることが示された。本試料の常温でのcritical doseは、一般的な有機化合物によく見られるようにわずか10-20e-nm-2であるため、電子損傷を最低限に抑えるためにdose rateを10 e-nm-2s-1と非常に低く設定し、total doseを10 e-nm-2(= 0.1 e-Å-2)に抑えた。このような非常に低い電子照射であっても高感度のCMOSカメラ(Gatan社製OneView)は、非常に明瞭な回折パターンを与える。既知の結晶形であれば電子回折パターンは容易に計算できるため、L-ヒスチジンの二つの異なる安定結晶形を明瞭に同定することができる(Fig. 3、Fig. 4)。また、測定時間は1分未満であり非常に高いスループットでの測定が可能である。本TEM測定には、日本電子製JEM-ARM200Fを用いた。
Fig.2

L-ヒスチジン塩酸塩一水和物の(a) TEM像および (b) 直径100 nmのナノビーム回折(NBD)パターン。NBDは(a)の白丸の範囲に照射した。(参考文献4より引用)
Fig.3
(a)L-ヒスチジン(orthorhombic)の実測電子回折パターン。(b)L-ヒスチジン(orthorhombic)の計算電子回折パターン、(c)L-ヒスチジン(monoclinic)の計算電子回折パターン。(参考文献4より引用)
Fig.4

(a)L-ヒスチジン(monoclinic)の実測電子回折パターン。(b)L-ヒスチジン(orthorhombic)の計算電子回折パターン、(c)L-ヒスチジン(monoclinic)の計算電子回折パターン。(参考文献4より引用)
一方で、13C CPMASと異なり、分子表面に多数存在する水素核を観測する1H NMRは、分子間パッキングに非常に敏感である。高速MASにより可能となる1H NMRの高分解能スペクトルをFig. 5に示す。Orthorhombic形とmonoclinic形でほとんど差が見られない13C CPMAS測定と異なり、明らかにパターンが変化していることが見て取れる。さらに1Hの天然存在比は99%以上であり、天然存在比がわずか1.1%の13Cと異なり容易に1H-1H原子間の相関測定が可能であり、分子間パッキングの違いをより明瞭に観測できる。1H DQ (double-quantum) / SQ (single-quantum)二次元相関NMRをFig. 6に示す。二つの結晶形で分子間相互作用が異なるため、明瞭なパターンの違いが見て取れる。たとえば、H1とH4の距離はorthorhombicでは4.1 Åであり相関を与えるには遠すぎるのに対して、monoclinicでは2.86 Åであり強い相関信号が観測される。距離の違いは1.4倍に過ぎないが、双極子相互作用は距離の3乗に比例するために相互作用の違いとしては2.9倍もの違いとなり、相関ピークの有無という形でNMRスペクトルに明瞭な違いを与える。また、1H DQ/SQ 二次元相関測定は数分から数十分程度で測定が可能であり、数時間から1日程度の測定時間を要する13C CPAMS測定と比べても高いスループットで結果が得られることは特筆すべきであろう。
Fig.5
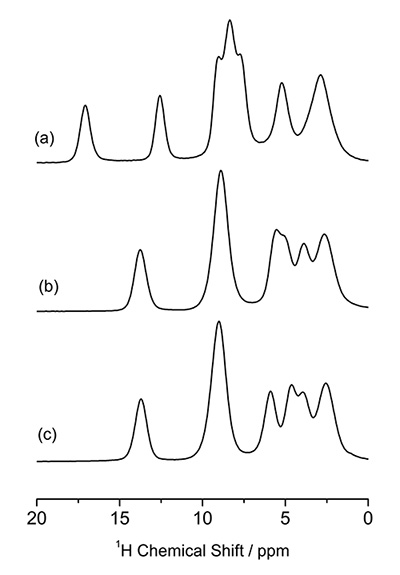
(a)L-ヒスチジン(orthorhombic)、(b)(monoclinic)、L-ヒスチジン塩酸塩一水和物の1H固体NMRスペクトル。(参考文献4より引用)
Fig.6
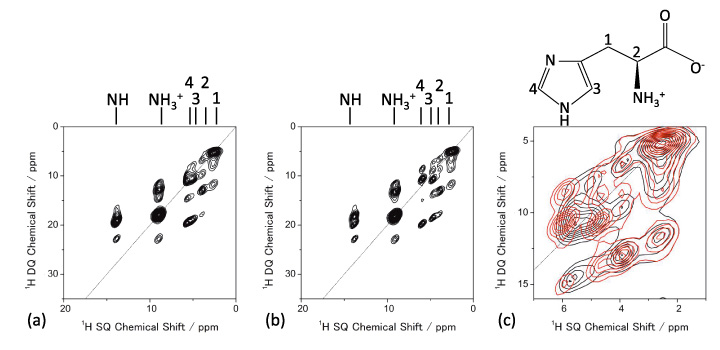
(a)L-ヒスチジン(orthorhombic)、(b)(monoclinic)の1H DQ/SQ相関固体NMRスペクトル。拡大した重ね合わせスペクトルを(c)に示した(黒: orthorhombic、赤: monoclinic)。(参考文献4より引用)
1H NMRにより得られるより面白い情報として、水素位置の決定があげられる。L-ヒスチジンのイミダゾール環の窒素はτ位とδ位のどちらかもしくは両方に水素原子がつきNHとなりうる。またZwitterionになりうる。これらの異性体の違いは、水素位置の違いのみであり、回折法により区別することは困難である。一方で、NMRでは直接水素核と窒素核の相関を観測することにより、これらを容易に区別することができる。1H/14N 二次元相関NMRスペクトルをFig. 7に示す。 L-ヒスチジン塩酸塩一水和物では、3つのNH相関信号が得られた。これは3つのNHの存在、すなわちτ位とδ位がともにNHになっていることを示している。一方で、すべてのスペクトルで14Nの周波数の低いところにひとつの相関信号が現れている。これは、NH3+の存在を示しており、いずれもZwitterionになっていることがわかる。このように1H/14N 二次元相関測定は、異性体の同定に非常に有用な情報を数分~数十分で与えるハイスループットの測定である。
以上の固体NMR測定には、JEOL RESONANCE製の外径1 mmの固体NMR試料管を用い、JNM-ECZ600R分光器を用いた。
Fig.7
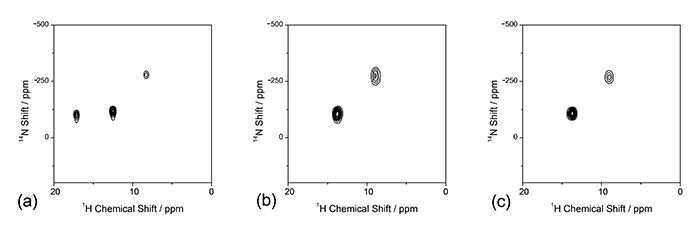
(a)L-ヒスチジン塩酸塩一水和物、(b)L-ヒスチジン(orthorhombic)、(c)L-ヒスチジン(monoclinic)の1H/14N相関固体NMRスペクトル。(参考文献4より引用)
上記結晶多形の測定のために新規開発した手法
窒素核には核スピンを有する二つの同位体、14Nと15Nが存在している。天然存在比では99%以上の窒素は14Nであるにもかかわらず、ほとんどすべての窒素NMR測定では天然存在比がわずか0.4%以下の15Nが選ばれ、低感度に苦しんでいる。これは、14Nのもつ整数スピンと四極子核という特徴が、14N NMR測定を困難にしているためである。われわれは、この問題を解決するために、高速MASのもとで1H/14N相関NMRを測定する手法を開発した[5]。この手法により、1 mg以下の極微量の試料から数分程度で1H/14N相関二次元NMRスペクトルを測定することに成功した。また、1H DQ/14N/1H SQ 三次元NMR測定などへの拡張も容易である[6]。さらに本アプローチは、14Nに限らず他の観測困難核にも適用できる。低分子医薬品によく見られる塩酸塩の35Cl NMR測定にも応用された[7]。
NMR信号の解釈には、分子構造と信号の帰属が必須である。二つの核スピンの間の相関を与える二次元相関NMR法は、信号帰属に重要な情報を与えるが連鎖帰属には3スピン間の相関を与える三次元NMRがより明瞭な情報を与える。われわれは13C/1H DQ/1H SQ 三次元相関NMR法を開発し1 mg以下の微量試料を用いて13C-1H-1Hの3スピンの相関情報からL-ヒスチジン塩酸塩一水和物の信号帰属に成功した[8]。13Cの天然存在比はわずか1.1%であるが、高速MASによる感度上昇を活用することにより天然存在比での低分子有機物の明瞭な信号帰属の道を開いた。
塩・共結晶[9]
APIの結晶は必ずしも望ましい性状(溶解度・安定性など)を示すとは限らず、無害なほかの成分と組み合わせることによる改善が幅広く試みられてきた。APIは多くの場合塩基性であり、酸性のcoformerから水素原子を受け取り、分子間イオン結合を生成させ塩が形成されることにより物性の改善が可能である。近年、APIとcoformerのpKaの差が3以下の場合にしばしば得られる共結晶と呼ばれる新しい形態の利用が広まりつつある(Fig. 8)。このとき、分子間の相互作用は水素結合や分子間力となり、水素原子の移動を伴わない。さらに塩と共結晶の中間であるcontinuumといった形態も報告されている。これらの形態の同定は、医薬品の開発のみならず知財の確保の上でも非常に重要である。一方で、pKaの差が3以下の時にはいずれの形態もとりうる上に、これらの形態の違いは水素原子の位置のみであるためX線回折から形態を同定することは困難である。一方で、固体NMRの視点でこれらの形態を読み解くと、水素核と窒素核の核間距離の違いとして明瞭な解を与えることができる。NMRでは、原子核間に双極子相互作用が存在し、その大きさは核間距離の3乗に反比例、それぞれの核の時期回転比に比例することが知られている。すなわち、水素核と窒素核の間の双極子相互作用の大きさを測定することにより、核間距離が得られ、塩/共結晶/continuumの明確な判別が可能となる。
Fig.8
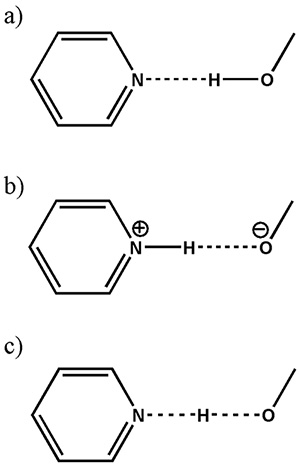
(a)共結晶、(b)塩、(c)continuumの模式図。(参考文献9より引用)
ここでは、単結晶X線回折および1H NMRから塩・共結晶問題に取り組む複合的アプローチを提案する。API/coformerのモデルとして4種類の試料を調製した(Fig. 9)。まず、これらの試料の単結晶X線回折から全体構造を決定した。この構造から明瞭に分子間相互作用が存在することが明らかになった。しかしながら、水素位置の決定は容易ではなく、測定に用いる装置や解析ソフトにより異なる値を示し、信頼できる位置を得ることができなかった。そこで、固体NMR法による1H-15Nの核間距離の決定を行った。単結晶X線回折により得られた構造から、窒素核は原子間相互作用に寄与しており1Hと15Nの核間距離を決定することにより、原子間相互作用の強さを見積もることができる。たとえば、SA-2の1H-15N距離は1.25 Åであり粉末X線回折により得られた分子間の窒素-酸素の距離2.54 Åと比較することにより、1Hは窒素と酸素の中間に位置しておりcontinuumであると同定した(Fig. 10)。SA-2は強い酸と強い塩基からなるpKaの差が3以上となる系であるにもかかわらず、塩ではなくcontinuumであったことは非常に面白い結果である。またこれは、pKaの差のみでは誤った結果につながり、固体NMR法による水素位置決定の重要さを強く示している。同様にしてこれら4つの試料の形態の決定に成功した(Fig. 10)。低い15Nの天然存在比と感度のため、残念ながら本NMR測定のスループットは限られている。下記に記す新規測定法を開発しスループットの改善を行ったが、それでもなお各サンプルそれぞれにおおよそ5日間の測定時間を要した。スループットの点からはまだ改善点が多いが、他の手法では得られない情報を得ることができるという点で価値のある測定と信じる。
本NMR測定には、JEOL RESONANCE製の外径1 mmの固体NMR試料管を用い、JNM-ECA700II分光器を用いた。
Fig.9

(a)SA1(3-nitrobenzoic acid and N,N-dimethypyridin-4-amine)、(b)SA2(3,5-dinitrobenzoic acid and 4-ethylpyridine)
(c)CO1(4-nitrobenzoic acid and 3-ethylpyridine)、(d)CNT1(pentachlorophenol and 4-methylpyridine)の構造。(参考文献9より引用)
Fig.10
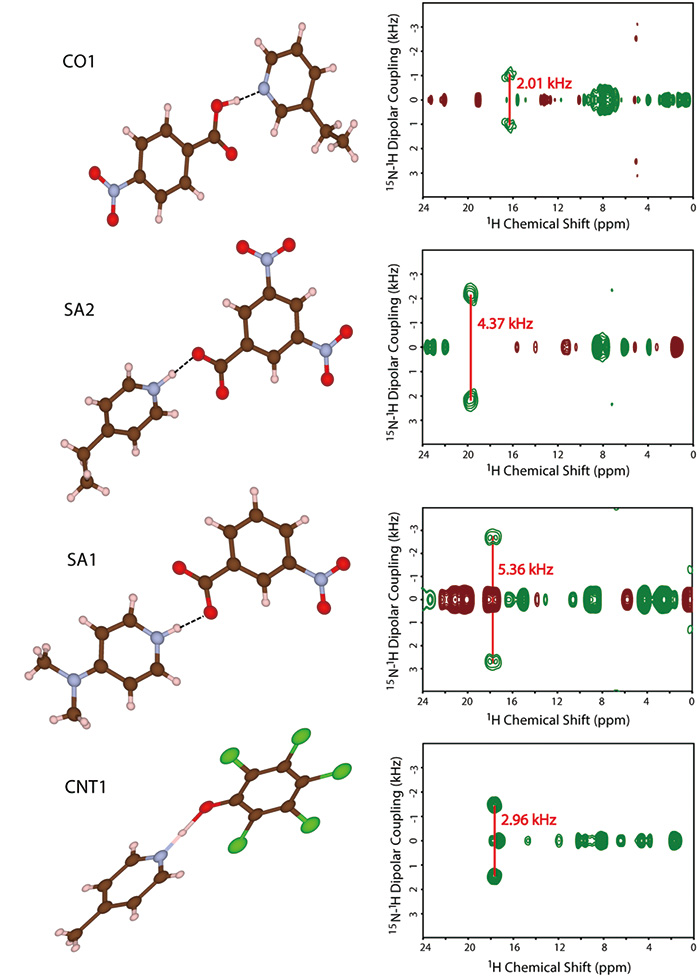
SA1, SA2, CO1, CNT1の分子構造と15N-1H距離測定。(参考文献9より引用)
塩/共結晶(新規開発した手法)
本手法では、1H-15Nの核間距離測定にあたり新規手法を開発した。15Nは小さな磁気回転比(1Hの約1/10)しか持たず、天然存在比もわずか0.4%以下である。そのため、1H-15Nは小さな双極子相互作用しか与えず、また感度は非常に低くなる。さらに深刻な問題として、観測される1H-15N双極子相互作用の大きさは、実験条件により左右されることが多く、信頼性の高い精密測定が困難であった。そこで、われわれは1H-15N双極子相互作用を高速MASのもとで測定するinv CP-VC法を開発した[10]。詳細は省くが、本手法は15N核を測定する代わりに1H核を測定することにより飛躍的に感度を向上させ、また、適切な実験条件を満たす核のみが信号に寄与することにより[11]、信頼性の高い精密測定が可能になった。また、必要になる試料量は1 mg以下であり、微量試料の測定に道を開いた。
NMR測定においては、測定前のスピン系が十分熱平衡に近い状態にある必要がある。このため、測定前に縦磁化緩和時間(T1)と同じオーダーの待ち時間を挿入する必要がある。一般には低分子有機物の1H T1緩和時間は数秒程度であることが多いが、安定性が非常に高い結晶である医薬品の1H T1緩和時間はしばしば100秒のオーダーもしくは、さらに長くなることが多い。このとき、測定の待ち時間は1スキャンあたり100秒のオーダーとなり、非常にスループットが低くなってしまう。さらに深刻なことに、本測定で測定を行うNHプロトンは物理的にもスペクトル的にも他の1Hから離れているため、特に高速MASのもとで他の1Hよりもいっそう長い1H T1緩和時間を示す。われわれは、このスループットの低下を緩和する手法を開発し本測定に適用した[12]。NHプロトンも低速MAS(< 20 kHz)のもとでは、すばやい1H-1Hスピン拡散のために他の1Hと同様のT1緩和時間を示す。しかしながら、高速MASのもとでは1H-1Hスピン拡散が抑制されるためNHプロトンの緩和時間が長くなる。そこで、待ち時間の間に1H-1Hスピン拡散を促進するRFDR(radio frequency driven recoupling)法を適用した。また1H-1Hスピン拡散の効率を最大化する位相回しを新規開発した[13, 14]。
結論 (まとめ、おわりに)
TEMやNMRを代表とする先端的測定機器の近年の技術革新には目を見張るものがあり、従来不可能であった測定が実現しつつある。TEMにおける高感度カメラは、クライオTEMの起爆剤となったが同様にあらゆる有機化合物の観測に有用である。NMRにおける高速試料回転技術は、これまでほとんど利用されてこなかった固体試料の1H NMRを実用的なものとした。これらの新しく得られる情報に加えて、これらの技術はスループットの改善につながっていることも実用的には見逃せない。さらに重要なことは、これらの技術を単独で用いるのではなく、得られる情報を適切に組み合わせることにより他のいかなる手法でも得られない重要な情報を効率的に得ることができる。日本電子株式会社で推進しているYOKOGUSHI戦略はまさにこの考えにのっとったものであり、本稿がそのよい具体例のひとつとなることを祈っている。
本稿では、X線回折・電子回折・固体NMRを有機的に組み合わせることにより、低分子医薬品で問題となっている結晶多形の問題および塩/共結晶の問題に解を与える試みを行った。X線回折・13C CPMAS NMRの組み合わせは、結晶形の同定に幅広く用いられているが、電子回折により製剤中に含まれるサブμm以下の微小結晶からも結晶形が同定できること、1H DQ/SQ NMRにより13C CPMAS NMRでは判別できない結晶形が同定できることを示した。さらに高速の試料回転によりのみ可能となる1H/14N相関NMR測定を用いることにより、水素位置を精密決定できることがわかった。塩/共結晶の同定には、水素位置を決めることによる分子間結合の理解が必須である。水素位置は単結晶X線回折法を用いても精密決定が困難であったが、単結晶X線回折による骨格構造の決定と固体NMRによる1H-15N距離の精密決定により、明確に塩/共結晶/continuumを区別できることが示された。非常に重要なことにpKaの差が3を超える組み合わせであっても塩とならずにcontinuumを形成していることが示された。
謝辞
本研究は、理化学研究所CLST-JEOL連携センターのメンバーと協力して遂行したものであり、メンバー各位に謝意を表するとともに、多大なるサポートをいただいた日本電子株式会社および株式会社JEOL RESONANCEの同僚たちに感謝する。また、多くの結果はAMES研究所、京都大学、東京農工大学、Warwick大学、 IISc Bangalore、Lille大学、Versailles大学、Michigan大学をはじめとする多くの共同研究の成果であり、ここに関係各位に深く感謝する。
参考文献
- Y. Nishiyama*, Fast Magic-Angle Sample Spinning Solid-State NMR at 60-100 kHz for Natural Abundance Samples, Solid State Nuclear Magnetic Resonance, 78 (2016) 24-36. DOI: 10. 1016/j. ssnmr. 2016. 06. 002.
- Y. Nishiyama*, Solid-state NMR under ultrafast MAS rate of 40 ‒ 120 kHz, in Experimental Approaches of NMR Spectroscopy ‐ Methodology and Application to Life Science and Materials Science, Springer (2017).
- T. Kobayashi, Y. Nishiyama*, M. Pruski*, Heteronuclear correlation spectroscopy with inverse detection, in Modern Methods in Solid-State NMR: A Practitionerʼs Guide, Royal Society of Chemistry (2018).
- T. Oikawa, M. Okumura, T. Kimura, Y. Nishiyama*, Solid-state NMR meets electron diffraction: Determination of crystalline polymorphs of small rganic microcrystalline samples, Acta Cryst. C73 (2017) 219‒228. DOI: 10. 1107/S2053229617003084.
- Y. Nishiyama*, Y. Endo, T. Nemoto, H. Utsumi, K. Yamauchi, K. Hioka, T. Asakura, "Very fast magic angle spinning 1H-14N 2D solid-state NMR: sub-micro-liter sample data collection in a few minutes", J. Magn. Reson. 208 (2011) 44-48.
- G. N. M. Reddy, M. Malon, A. Marsh, Y. Nishiyama, S. P. Brown*, A fast magic-angle spinning three-dimensional NMR experiment for simultaneously probing H-H and N-H proximities in solids, Analytical Chemistry, 18 (2016) 6209-6216.
- M. Malon, M. K. Pandey, Y. Nishiyama*, Revealing the Local Proton Network Through Three-Dimensional 13C/1H Double Quantum/1H Single Quantum and 1H Double Quantum/13C/1H Single Quantum Correlations Fast MAS Solid-State NMR Spectroscopy at Natural Abundance, J. Phys. Chem. B 121 (2017) 8123-8131. DOI:10. 1021/acs. jpcb. 7b06203.
- L. Rajput†, M. Banik†, J. R. Yarava, S. Joseph, M. K. Pandey, Y. Nishiyama*, G. R. Desirajua*, Exploring the salt-cocrystal continuum with solid-state NMR using natural abundance samples: implications for crystal engineering, IUCrJ 4 (2017) 466-475. DOI: 10. 1107/S205225251700687X.
- Y. Nishiyama*, M. Malon, M. J. Potrzebowski, P. Paluch, J. P. Amoureux*, Accurate NMR determination of C‒H or N‒H distances for unlabeled molecules, Solid State Nucl. Magn. Reson. 73 (2016) 15-21. DOI: 10. 1016/j. ssnmr. 2015. 06. 005.
- P. Paluch, J. Trébosc, Y. Nishyama, M. Potrzebowski*, M. Malon, J. -P. Amoureux*, Theoretical study of CP-VC: a simple, robust and accurate MAS NMR method for analysis of dipolar C-H interactions under rotation speeds faster than ca. 60 kHz, J. Magn. Reson. 252 (2015) 67-77.
- Y. Q. Ye, M. Malon, C. Martineau, F. Taulelle, Y. Nishiyama*, Rapid measurement of multidimensional 1H solid-state NMR Spectra at ultra-fast MAS frequencies, J. Magn. Reson. 239 (2014) 75-80.
- Y. Nishiyama, R. Zhang, A. Ramamoorthy, Finite-Pulse Radio Frequency-Driven Recoupling with Phase Cycling for 2D 1H/1H Correlation at Ultrafast MAS Frequencies, J. Magn. Reson. 243 (2014) 25-32.
- R. Zhang, Y. Nishiyama, P. Sun, A. Ramamoorthy*, Phase Cycling Schemes for finite-pulse-RFDR MAS Solid State NMR Experiments, J. Magn. Reson. 252 (2015) 55-66.