単粒子解析
単粒子解析
single particle analysis
[目次:理論(電子の散乱/回折/結像)]
タンパク質や核酸など生体高分子の水溶液薄膜を微小孔カーボン薄膜中に形成させ、急速凍結してタンパク質分子等を非晶質の氷薄膜に包埋し、氷薄膜中に分散した分子(単粒子)像を液体窒素温度にて撮影した電子顕微鏡画像から、画像解析によってその三次元構造を解析する手法。
撮影した画像にはタンパク質粒子を様々な方向から投影した像が記録されている。それぞれの粒子像に画像面内での回転と平行移動を施して、外形や密度分布が同じ粒子像を集めてグループ分けする。その後、各グループの粒子像を加算平均して像のS/N比を向上させる。各グループの粒子像の投影方向のオイラー角(粒子が画像面と成す角)を推定し、その角度に沿って逆投影することによって、タンパク質の三次元構造を再構成する。
試料作製法や解析ソフトウエアの発展、ならびにCMOSを用いた直接露光検出器の登場により、単粒子解析法の高分解能化・実用化が進んだ。極めて少量(密度: 数mg/mL、容量: 数µL/グリッド)のサンプルでの解析が可能で、結晶化の困難なタンパク質の構造解析にも有効である。2018年現在、最高到達分解能は0.16 nmである。
試料作製および電子顕微鏡観察法
ほぼ単一の構造の高純度のタンパク質粒子を水溶液に分散させる。タンパク質の構造を損なわないようにするために急速凍結(-180°C以下)により氷包埋する。タンパク質は C、H、O、N、S など軽元素で構成され電子顕微鏡像のコントラストが低いため、試料作製の際、タンパク質粒子を取り囲む氷の厚みを十分薄くすることが重要である。
凍結した状態で試料を電子顕微鏡内に挿入する。電顕像の撮影は液体窒素温度で行う。電子線照射による試料の損傷を抑えるため、観察の際は照射電子線量を低くする必要がある。軽元素からなるタンパク質粒子は散乱コントラストによる観察が困難なため、位相コントラストによって観察する。その際、低周波領域のコントラストを向上させるため、対物レンズを大きく(-0.5~数µm)デフォーカスさせる。
画像取得
低電子線量で撮影されたタンパク質粒子像のS/N比は極めて低い。S/N比の高い粒子像を得るために、数百から数千枚の画像を撮影し、画像を分類し揃えた後で数万から数十万の粒子像を抽出し加算平均する。近年は自動画像取得システムを使って、数日間にわたって数千枚の画像を撮影する。
電子線照射によるタンパク質粒子のドリフトは単粒子解析の分解能低下の重要な一因である。シンチレーターを介さない直接露光検出器を用いた単電子検出により、電顕像自体の分解能が向上した。さらに高フレームレート(5~10フレーム/秒)で動画撮影することで粒子のドリフトの影響が軽減し、撮影後に各フレーム間での粒子像の動きを補正することで、高解像度タンパク質粒子像の取得が可能になっている。
画像解析
粒子自動検出ソフトにより、数千枚の電子顕微鏡画像から数万〜数十万のタンパク質粒子像を自動抽出する。粒子像は様々な方向で投影されているので、抽出した画像を回転並進させ、同じ方向の投影像に分類した上で位置と向きを揃え、各分類画像を平均化することでS/N比の高い100~200個程度のタンパク質粒子像を得る。この段階では、粒子像の三次元的な方向(オイラー角、今の場合、画像面と成す角)は定まっていない。適当な方向を仮定した粒子の初期モデルを与えて、その投影像と得ている画像との比較から各粒子像のオイラー角を推定する。推定したオイラー角を付与した粒子像を逆投影によって初期の三次元構造を得る。その三次元構造から再び投影像を作成する。再投影した像と元の画像とを比較してオイラー角を再付与する。この画像を逆投影して次のステップの三次元構造を得る。得られた三次元構造が収束するまでこの操作を繰り返す。計算の各段階では統計学の手法により計算結果を評価し、最終的に最も確からしい三次元構造を得る。
他の解析手法との比較
電子顕微鏡を用いた構造解析法には、電子線結晶構造解析法、電子線トモグラフィー、単粒子解析法がある。電子線結晶構造解析法では、二次元結晶あるいは薄い三次元結晶を用いて、X線構造解析と類似の方法で構造解析する。分解能は0.2 nmを切っている。電子線トモグラフィーでは、一つの試料を電子顕微鏡内の試料ホルダーで連続的に傾斜して、さまざまな角度からの投影像を撮影し、得られた画像を逆投影して三次元構造を得る。この方法では、加算平均による画像のS/N比の向上を実施することはできない。生体物質への応用では、細胞レベルでの機能構造の解析に用いられており、分解能は数〜10 nmオーダーである。しかし、得られたトモグラムに同じタンパク質粒子が数多く含まれる場合はサブトモグラム平均が可能で、この方法による分解能向上は0.3 nmに近い領域に到達している。
従来から用いられているタンパク質の構造解析法は、X線結晶構造解析法ならびに核磁気共鳴法(NMR)である。前者ではサイズが10 µm以上の結晶が必要であり、結晶化できないタンパク質は構造解析できない。NMRでは解析できる分子量が~5万以下という制限がある。単粒子解析法にはこれらの制限はなく(ただし、分子量10万以下の小さな分子では画像のS/N比が悪く現時点での課題)、分解能はX線結晶構造解析と同程度に近づいており、単粒子解析法の発展と利用が期待されている。
(データ提供: 大阪大学 難波啓一特任教授、加藤貴之特任准教授)
An electron microscopy method to obtain the three-dimensional (3D) structure of biological macromolecules, such as proteins and nucleic acids, by analyzing electron microscope images of the macromolecules (proteins, etc.) recorded as dispersed particles (single particles). A solution containing a protein is made into a thin film on a holey carbon film attached on an EM grid and is then rapidly frozen so that the particles are embedded in an amorphous ice film. TEM images are taken at Liq.N2 temperature, and the 3D structure is reconstructed by image processing and analysis.
A number of projection images of the 3D structures of protein particles in various orientations are recorded in the acquired Cryo-TEM images. Each particle image is rotated and translated in the image plane to collect the images having the same external shape and the same density distribution to sort them into groups of different 3D orientations. Then, the images in the same groups are added and averaged to improve the signal-to-noise ratio of the images. Next, the Euler angles of the projection orientations (angles of the particles against the image plane in the present case) are estimated. Finally, the 3D structure of the protein is reconstructed by back projection of these particle images along the estimated Euler angles.
Single particle analysis has been dramatically improved in its achievable resolution and practical application by the advent of a CMOS direct electron detector, together with innovations in specimen preparation techniques and analysis software programs. The method is applicable to an extremely small amount of sample solution (protein concentration: several mg/mL, solution volume: several µL/grid), and is especially effective for structural analysis of proteins that are difficult to crystallize. As of 2018, the highest resolution achieved with the method is 0.16 nm.
Specimen preparation and observation method
A highly purified protein solution is prepared in which protein particles with a homogeneous structure are dispersed. The protein particles are ice-embedded by rapid freezing (at –180 °C or less) so as to preserve their structures. It is important that the ice film embedding the protein particles must be prepared as thin as possible to produce high contrast TEM images because proteins are composed of light elements (C, H, O, N, S, etc.) and produce low contrast against the ice film by a small difference between their densities.
The specimen grid is inserted into the microscope column with its frozen state kept. TEM images of the specimen are recorded at Liq.N2 temperature. To suppress the damage to the specimen due to electron-beam irradiation, the electron dose onto the specimen must be set low. The specimen is observed using phase contrast because protein particles consisting of light elements are difficult to observe using scattering contrast. For observation of protein particles, a large amount of defocus of the objective lens (–0.5 to several µm) is used to enhance the image contrast, particularly in a low spatial frequency range to allow the detection and orientation determination of the particles.
Image acquisition
Images of protein particles recorded at a low electron dose have a very low-signal-to-noise ratio (S/N). To obtain the particle images with a high S/N, hundreds to thousands of Cryo-TEM images are collected, from which several tens of thousands to several hundreds of thousands of particle images are extracted and added and averaged after image classification and alignment. In recent years, several thousands of Cryo-TEM images are automatically acquired over several days using an automatic image acquisition system.
It is emphasized that the electron-beam-induced drift of ice-embedded protein particles is the main cause of degrading the resolution of the TEM image in the single particle analysis method. Extremely high frame rate of direct electron detectors (scintillator not used) that allow single electron counting and movie-mode image recording has improved the resolution of the TEM image. Recording a TEM image in the movie mode at a frame rate of 5 to 10 frames/sec allows the particle image drift to be greatly reduced by post image processing, namely motion correction of the particle images between the frames enables acquisition of high-resolution protein particle images.
Image analysis
From several thousands of the Cryo-TEM images, several tens of thousands to several hundreds of thousands of the protein particle images are automatically extracted using a particle detection software program. Since the particle images are projected in various orientations, the extracted images are rotated and translated so as to sort them out into many classes of the projection images in the same orientations. Then, the images sorted in each class are aligned in their positions and orientations and are averaged. As a result, 100 to 200 protein particle images with a high S/N are obtained. In this step, the 3D orientations of the particle images (Euler angles or the angles of the particles against the image plane in the present case) are not yet determined.
To obtain the Euler angles, first an initial particle model with arbitrary orientations is given. Secondary, the projection images of the model are produced. The Euler angle of each particle image is estimated by a comparison of the projection images and the TEM image. The particle TEM images with the estimated Euler angles are subjected to back projection to produce an initial 3D structure of the particle. Then, this 3D structure is subjected to projection again to create the projection images. The re-projected images and the particle TEM images are compared to estimate the Euler angles again. These particle images are back-projected to obtain a better 3D structure in the next step. These steps are repeated until the obtained 3D structure is converged. In each step of calculations, the results are evaluated by statistical approaches. Finally, the most probable 3D structure is obtained.
Comparison with other analytical methods
Structural analysis methods using electron microscopy include Electron Crystallography, Electron Tomography and Single Particle Analysis. In Electron Crystallography, a 2D or thin 3D crystal is prepared and analyzed using techniques similar to those of X-ray Crystallography. Electron Crystallography provides a resolution better than 0.2 nm. In Electron Tomography, one specimen is placed in the specimen holder in the microscope column, and is subjected to serial tilt-series imaging at various angles. Then, back projection is applied to the acquired projection images for obtaining the 3D structure. In the method, it is not possible to improve the signal-to-noise ratio and the resolution of the images by averaging. For biological applications, the method is used for analysis of functional structures at the cell level, its resolution being on the order of several nm to 10 nm. But when the tomogram contains many particles of the same proteins, the use of the sub-tomogram averaging method improves the resolution close to 0.3 nm.
Conventionally-used structural analysis methods of proteins are X-ray Crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. The former method requires a crystal with a size of 10 µm or more. Thus, the method cannot be applied to proteins that are unable to crystallize. The latter method can be used only for proteins with a molecular weight of ~50,000 or less. However, Single Particle Analysis does not have such restrictions and its resolution has been becoming as high as that of X-ray Crystallography, although at present it is still difficult to apply this method to proteins with molecular weight less than 100,000 because of low S/N of their TEM images. In the future, the advancement and the more use of Single Particle Analysis are highly expected.
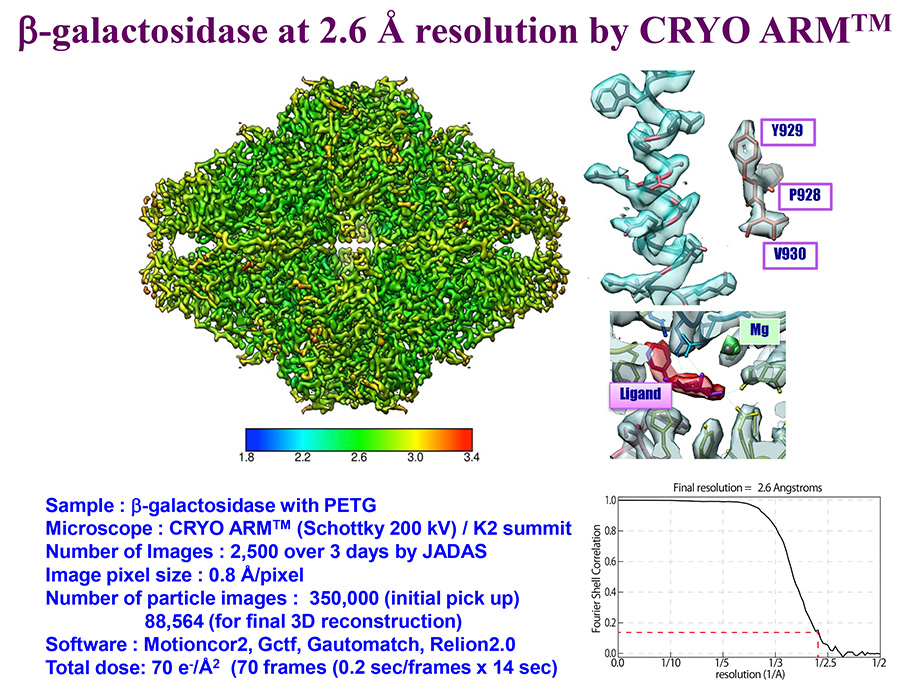
Data courtesy: Specially Appointed Professor Keichi Namba and Specially Appointed Associate Professor Takayuki Kato, Osaka University
関連用語から探す
説明に「単粒子解析」が含まれている用語





